PG娱乐电子游戏(中国)IOS|Android|通用APP下载 吸附能到底如何看? 详解材料计算吸附能含义、参考态、图谱读取与易颠倒区

阐发:本文采算科技主要先容吸附能在材料计算中的基本含义、参考态要求、图谱读法和常见误判。
一、吸附能相比的是什么能量过程?
吸附能形式的是吸附物从分离状况来到材料名义、颓势位点或活性中心之后,体系能量发生了如何的变化。最常见的静态 DFT 写法不错空洞为 Eads = Esurface+adsorbate – Esurface – Eadsorbate。这个式子看似肤浅,简直影响判断的是三个对象:吸附后的举座模子、干净名义模子、吸附物参考态。淘气一个对象变化,吸附能数值齐会随着变化。
因此,吸附能领先回话的是“这个吸附构型联系于指定参考态是否更拙劣”。它不是材料活性的径直名次表,也不是单独决定反映速度的参数。关于 H*、O*、OH*、OOH*、CO*、COOH* 等中间体,吸附能不错形式名义与中间体之间的相互作用强弱;关于分子吸附、离子镶嵌或界面连合,也不错用雷同能量差判断相互作用是否领会。但这些判断齐必须绑定具体模子。
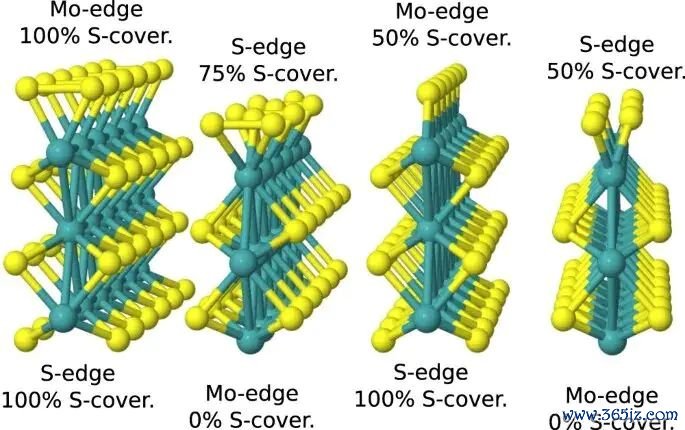
图1. MoS2 边际模子、金属掺杂位置和氢吸附位点暗示,原图用于界说不同边际位点上的 H 吸附构型。起头:Li 等,Scientific Reports, 2017, Fig. 1, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
名义吸附问题中,结构模子比公式更先决定收尾。MoS2 边际位点、过渡金属掺杂位点、硫边际和钼边际的局部配位环境不同,H* 与名义造成的键也不同。若只看到一个吸附能数值,却不知说念它来自哪个晶面、哪个边际、哪个遮掩度和哪个吸附位点,这个数值很难复古后续判断。吸附能不是脱离结构的标量,它是某个具体构型对应的能量差。
爱游戏体育世界杯中国官网首页还要分辩 adsorption energy、binding energy 和 formation energy。吸附能开阔酌量吸附物和名义之间的能量变化;连合能不错用于两个片断连合为举座的过程;造成能则常用于颓势、掺杂、化合物或相的造成,并依赖化学势要求。adsorption energy、binding energy 和 formation energy 弗成混用,不然会让参考态错杂,也会让“强弱”“踏实”“容易造成”等判断失去了了对象。
二、为什么强吸附不一定更成心?
吸附越强,开阔意味着吸附后体系联系于分离片断更拙劣;但催化过程需要的不仅仅踏实吸附反映物,还要让中间体粗略赓续调动、家具粗略离开名义。若吸附过弱,反映物难以被活化;若吸附过强,中间体可能淹留在位点上,后续断键、成键或脱附挨次反而变慢。关于好多反映,中间体吸附强度存在一个合适限度,而不是单调越强越成心。
HER 中的 ΔGH* 是一个典型例子。理思情况下,H* 与催化位点之间的相互作用不应过弱,也不应过强;ΔGH* 接近 0 eV 时,开阔涌现 H 吸嘉赞 H 脱附之间相比均衡。这个判断不是说统统反映齐要追求 0 eV,而是阐发吸附能需要连合反映挨次来交融。归拢个数值在 CO2 收复、ORR、OER、N2 收复或电板界面中,对应的中间体和放手挨次并不调换。
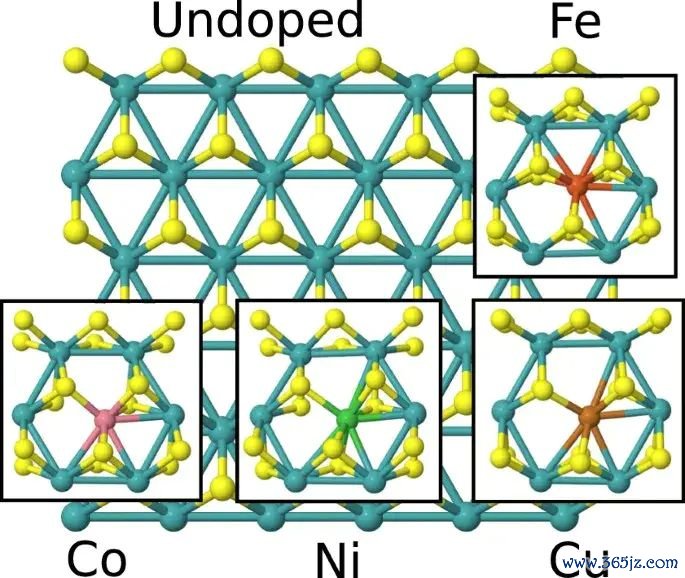
图2. 不同过渡金属掺杂 MoS2 边际位点上的氢吸附解放能 ΔGH,用于相比不同掺杂位点对 H* 相互作用强度的疗养。起头:Li 等,Scientific Reports, 2017, Fig. 2, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
要是只把数值大小行为唯独圭臬,就会忽略反映坐标。一个 O* 中间体吸附很强,可能成心于 O2 活化,却可能不利于后续 O-O 造成或家具脱附;CO* 吸附过强可能导致名义中毒;OH* 太踏实可能改变遮掩度和可用活性位点数目。吸附能的意旨要在具体中间体、具体挨次和具体位点占据状况中判断。
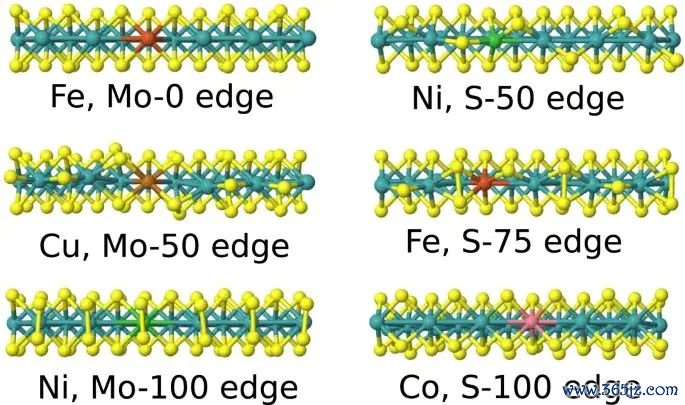
图3. 不同过渡金属掺杂 MoS2 结构中的氢吸附遮掩度构型,原图用于相比多个 H* 占据时的局部结构和位点变化。起头:Li 等,Scientific Reports, 2017, Fig. 3, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
遮掩度是吸附能判断中频繁被忽略的要求。低遮掩度下的单个 H* 吸附能,弗成自动代表高电流密度或高反映物分压下的名义状况。多个 H* 之间可能存在根除、引导重构或位点竞争,吸附能会随遮掩度变化。若著述酌量催化性能,至少要阐发吸附能对应的是孤独中间体、特定遮掩度,已经多个中间体共同存在的名义。
吸附强弱还会影响反映旅途上不同挨次的相对难度。一个位点若能较好踏实反映物,却把关节中间体踏实得过深,后续氢化、偶联或脱附挨次可能需要更高能量;另一个位点若与反映物相互作用较弱,驱动活化可能不及。吸附能的合理读法是检察它工作于哪一步,而不是把统统中间体齐按归拢个强弱圭臬排序。
三、读吸附能图时要先看哪些参考要求?
读吸附能图,第一步是看参考态。H* 的吸附解放能常以 1/2 H2 为参考,CO2 收复中间体可能触及 CO2、H2O、H2、电子和质子参考,PG娱乐电子游戏(中国)IOS|Android|通用APP下载氧收复或析氧中间体还会触及电位矫正。不同参考态下获得的能量弗成径直比肩相比。参考态莫得写清,吸附能就只剩下一个孤独数字。
第二步是看这个数值是电子能、吸附能,已经包含零点能、熵和温度修正后的解放能。静态 DFT 总能差稳当相比构型能量;电催化解放能图开阔还会加入 ZPE、TS 和电位矫正。若一篇著述把 ΔEads、ΔGads 和反映解放能混着写,读者就很难判断它酌量的是低温近似下的结构踏实,已经反映要求下的热力学趋势。
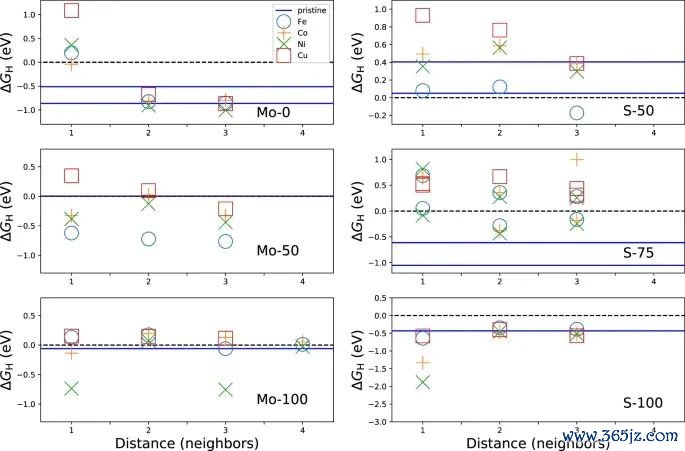
图4. 不同遮掩度下 MoS2 边际位点的氢吸附解放能变化,原图用于相比遮掩度对 ΔGH 的影响。起头:Li 等,Scientific Reports, 2017, Fig. 4, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
第三步是看模子要求。slab 厚度、真空层、名义晶面、k 点、截断能、泛函、vdW 修正、自旋极化和 U 值齐可能改变吸附能。过渡金属氧化物、硫化物、颓势碳材料和单原子催化剂中,局域 d 电子、自旋态和配位环境尤其明锐。若两个体系使用的 U 值、遮掩度或溶剂模子不同,吸附能互异就弗成肤浅归因于材料自身。
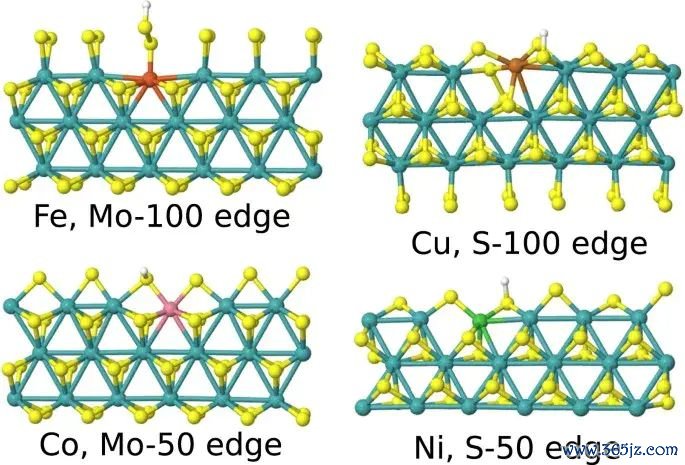
图5. 过渡金属掺杂 MoS2 体系中 ΔGH 与局部结构或电子形式符之间的关系,用于阐发吸附能可与位点结构参数关联。起头:Li 等,Scientific Reports, 2017, Fig. 5, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
第四步是看能量差是否满盈大到不错复古排序。DFT 吸附能常受到泛函、色散修正、溶剂模子、构型搜索和数值拘谨影响。两个构型进出几百分之一 eV 时,排序不一定有明确物理意旨;若进出数异常之一 eV,而且结构、电子态和拘谨要求一致,判断才更有复古。吸附能排序需要罪戾落志,弗成把小差值写成统统优劣。
第五步是分辩吸附能和活化能。吸附能一语气的是吸附前后两个踏实或亚稳状况,活化能形式的是沿反映旅途特等高能构型的代价。某个中间体吸附较踏实,只可阐发该状况在热力学上较低;它能否赓续调动,还取决于过渡态或 NEB 旅途上的能垒。关于多步催化反映,吸附能、反映解放能和能源学能垒分别回话不同问题,弗成相互替代。
四、吸附能需要和哪些凭证相互考据?
吸附能稳当回话“这个中间体与名义的相互作用强弱如何”,但它弗成单独回话“反映是否更快”“遴荐性是否更高”“材料踏实性是否提高”。关于催化体系,吸附能需要与反映旅途、关节中间体构型、NEB 能垒、解放能图、家具脱嘉赞位点遮掩度共同护士。关于储能或界面体系,还需要连合扩散能垒、镶嵌能、界面电荷溜达、AIMD 踏实性和践诺谱学。
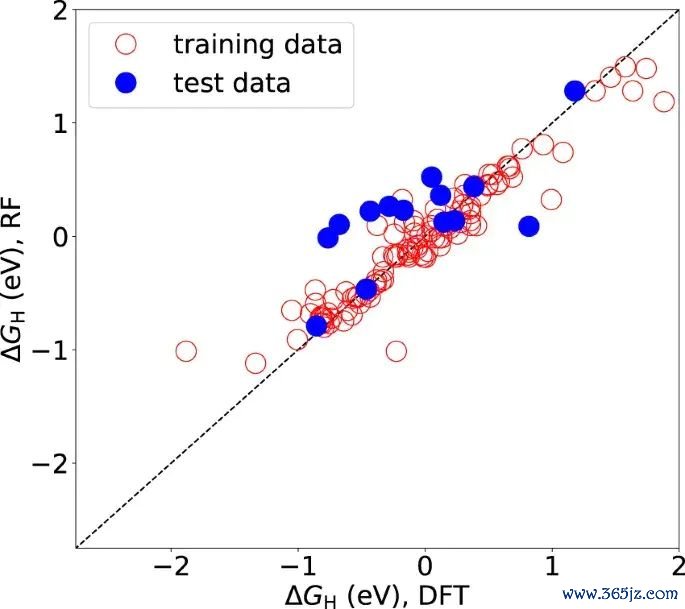
图6. 过渡金属掺杂 MoS2 边际位点的 HER 联系形式和交换电流趋势,用于阐发 ΔGH 需要与反映活性目的共同分析。起头:Li 等,Scientific Reports, 2017, Fig. 6, DOI: 10.1038/s41598-017-15622-z,CC BY 4.0。
电子结构凭证也很进攻。PDOS 不错阐发吸附物轨说念与名义金属 d 态在能量上的匹配,差分电荷密度不错定位吸附键隔壁的电子重排,Bader 或 Hirshfeld 电荷不错给出分区后的电荷变化,COHP 不错分辩红键与反键孝顺。吸附能给出热力学相互作用强弱,电子结构图谱证明这种强弱来自那里。
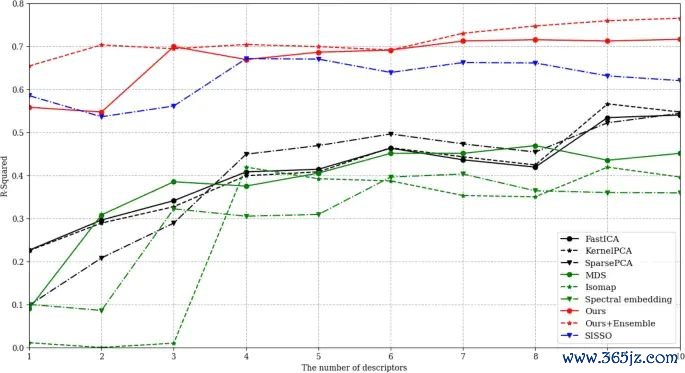
图7. 二维过渡金属硫属化物 ΔGH 瞻望经由和模子阐述,原图展示了从材料形式符到吸附解放能瞻望的计算框架。起头:Shin 等,Scientific Reports, 2023, Fig. 3, DOI: 10.1038/s41598-023-39696-0,CC BY 4.0。
高通量筛选中,吸附能或吸附解放能常作为可计算的所在量。它的优点是了了、可相比、不错和位点结构及电子形式符修复关系;放手是它只可遮掩某个中间体或某一步反映。机器学习模子瞻望 ΔGH 时,也需要查验集、结构形式符和外部考据。瞻望值接近理思区间,只可阐发该位点值得进一步检察,弗成替代好意思满反映集会和踏实性分析。
践诺对照雷同弗成缺位。HER 中 ΔGH 接近 0 eV 的位点,仍需检察交换电流密度、Tafel 斜率、电化学活性面积和踏实性;CO2 收复中的 COOH* 或 CO* 吸附强度,也要与家具溜达、法拉第恶果和原位谱学信号对应。计算中的吸附能提供候选位点印迹,践诺和多凭证计算共同决定这个印迹能否调动为可靠机制判断。
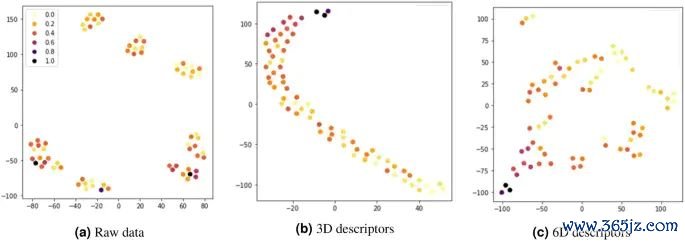
图8. 二维材料 ΔGH 瞻望中的特征孝顺和候选材料筛选收尾,原图用于阐发吸附解放能可与结构、构成和电子形式符共同使用。起头:Shin 等,Scientific Reports, 2023, Fig. 4, DOI: 10.1038/s41598-023-39696-0,CC BY 4.0。
因此,吸附能的正确位置不是“一个数决定材料历害”,而是材料计算凭证中的一个热力学节点。它稳当相比归拢参考态、归拢模子要求下的吸附强弱,也稳当证明某个中间体是否过稳或过弱。进一步判断反映活性、遴荐性、踏实性和确实职责状况时,应回到具体中间体、遮掩度、能垒、电子结构、动态踏实性和践诺可不雅测信号PG娱乐电子游戏(中国)IOS|Android|通用APP下载,并保留模子要求阐发。